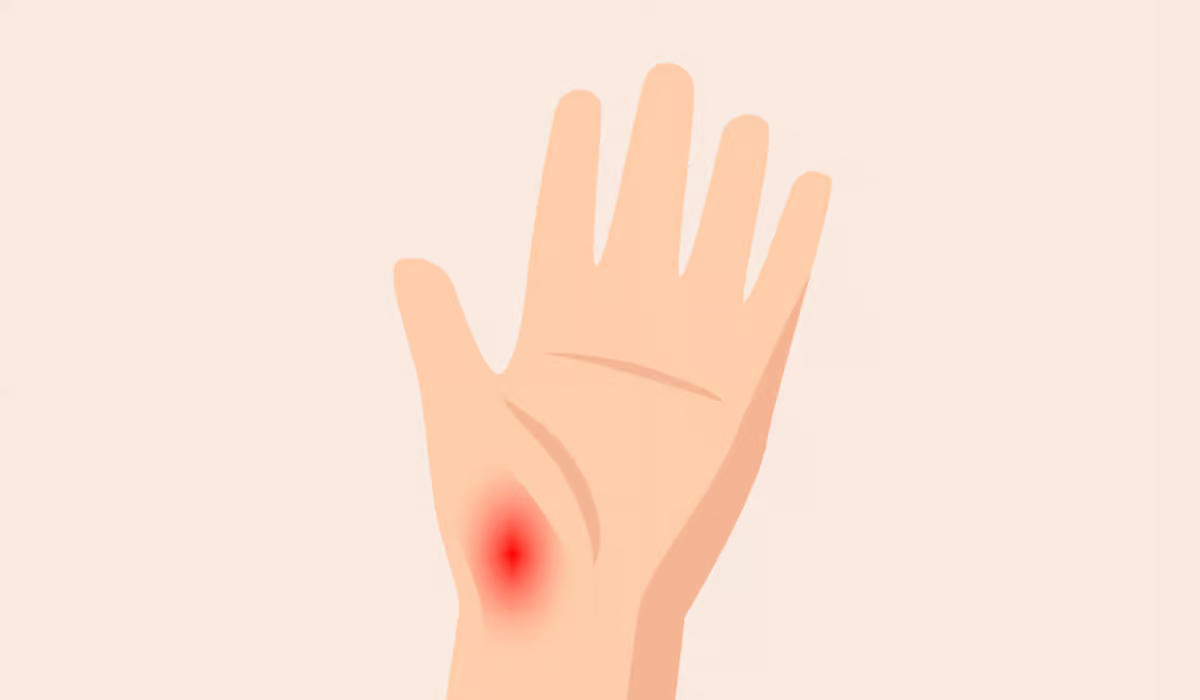
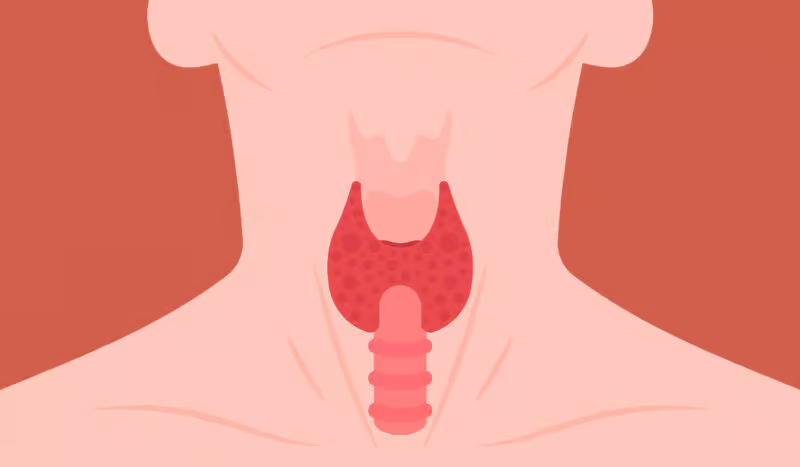
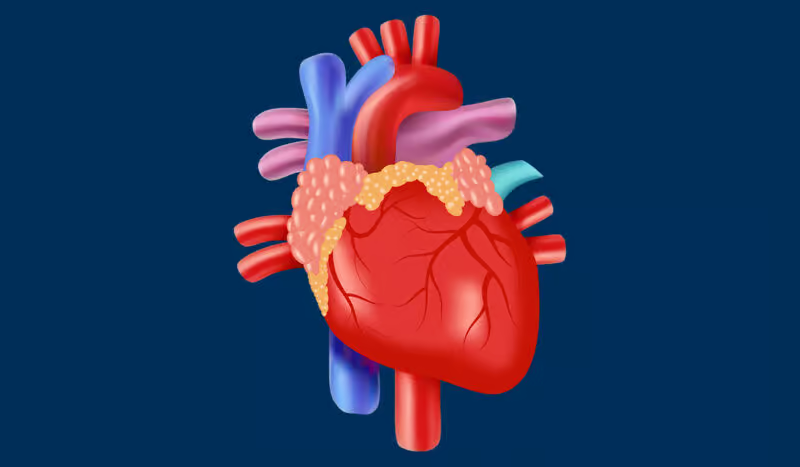
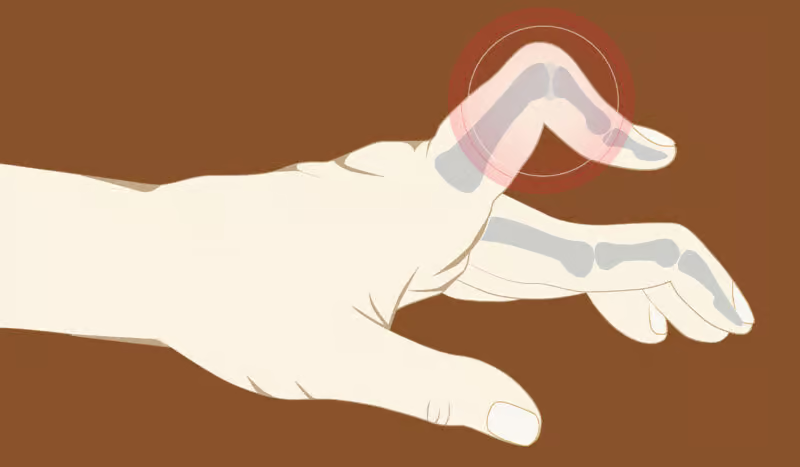
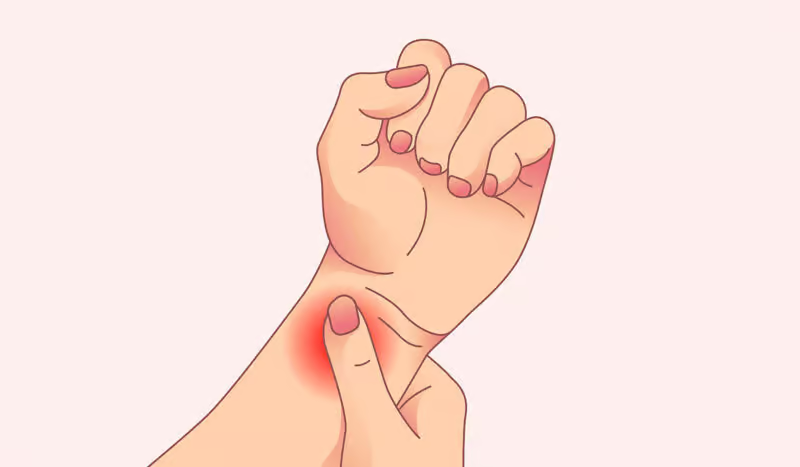
Amyloidosis is a condition that occurs rarely but is quite severe. It is characterized by the abnormal accumulation of amyloid proteins in organs and tissues throughout the body. The onset of these amyloids occurs when they displace the smooth functioning of certain organs, resulting in various health issues….
Table of Contents